*CUANDO EL CEREBRO SE CONVIERTE EN UNA ESPONJA SE CONVIERTE ASÍ SON LAS ENFERMEDADES PRIÓNICAS: "LA EVOLUCIÓN ES MUY RÁPIDA, EN MESES *
LA VOZ DE LA SALUD

Jesús Rodríguez Requena, investigador líder del grupo de enfermedades por priones del Instituto de Investigación Sanitaria de Santiago de Compostela (IDIS), compara las encefalopatías espongiformes con el alzhéimer, ambas de carácter neurodegenerativo, para ayudar a su comprensión. «Son parecidas, pero también se diferencian en muchas cosas. Las enfermedades por priones son mucho más rápidas. Por ejemplo, una persona que tenga alzhéimer, comienza a tener signos clínicos y pasan muchos años hasta que se alcanza la fase terminal. En las patologías priónicas son unos meses o poco más de un año». El causante de las mismas es un prión, una proteína mal plegada que es capaz de transmitir esta forma anómala a otras variedades de la misma, que se dirige al cerebro y se replica a lo largo del sistema nervioso. «La proteína priónica segrega y adquiere una forma anómala. Comienzan a formar unos agregados que causan daño. Suele ser de una forma espontánea, sin que se sepa muy bien el porqué», añade.
Rodríguez sabe que la población conoce una variante de estas encefalopatías espongiformes que se da en animales. «Los lectores estarán pensando en los priones de las vacas locas. Eso fue una cosa muy peculiar y puntual que ya no se produce». El primer caso español de encefalopatía espongiforme bovina se identificó en Lugo en noviembre del 2000. Según datos del Ministerio de Agricultura, Pesca y Alimentación, en el 2001 el número de focos se incrementó a 81; en el 2002, 127; en el 2003, 167 (el pico más alto de contagios); en el 2004 empezaba a descender: 137; y en el 2005, 98. Hasta el 2024, que se cerró con cero focos.
La forma en humanos del «mal de las vacas locas» es la enfermedad de Creutzfeldt-Jakob. En España se contabilizaron cinco víctimas mortales a raíz de este suceso. El primero, según el Instituto de Salud Carlos III de Madrid (ISCIII), se detectó en el 2005 en Madrid y durante el 2007 y el 2008 se diagnosticaron otros cuatro, tres en Castilla y León y otro en Cantabria. «Se produjo contaminación con priones que estaban en vacas infectadas. Lo ingeríamos y esos priones llegaban a nuestro cerebro», contextualiza. Si bien, remarca que estas enfermedades no son contagiosas. «Se podría pensar que hay que evitar a las personas que las padecen. No, lo que está pasando en su cerebro no es de ninguna manera contagioso». Además, esta forma de transmisión ya no se produce.
Origen esporádico, genético y adquirido
La incidencia de las enfermedades priónicas es de un caso por millón de habitantes y año. Por lo tanto, se estima que en Galicia se dan de dos a tres diagnósticos de este tipo al año, aproximadamente. Las formas más comunes en humanos son la enfermedad de Creutzfeldt-Jakob y el insomnio familiar fatal —aunque también se han estudiado algunos casos de síndrome de Gerstmann-Sträussler-Scheinker—. El origen de la primera es, sobre todo, esporádico o espontáneo. Es decir, se desconoce la causa que lleva a ese mal plegamiento proteico que inicia el proceso que lleva a la enfermedad. Aunque también puede existir algún caso genético.
En cambio, el origen del insomnio familiar fatal es, en la amplia mayoría de casos, genético, si bien pueden darse también casos esporádicos. La enfermedad está causada por mutaciones en la proteína responsable de la enfermedad. Existe otro tipo de desarrollo, el adquirido a través de la práctica médica: una intervención que, por desconocimiento, llevaba a la infección; en trasplantes, por ejemplo. Actualmente, al igual que el origen infeccioso, ya no se describen enfermedades priónicas con estos orígenes en humanos.
«Las formas más frecuentes son las espontáneas o esporádicas en las que se desconoce la causa. Esas corresponden con un 80 % de los casos. Luego están las genéticas que son menos, un 15 a un 20 %, y que afectan a una serie de familias», afirma el investigador del IDIS. Sobre estos últimos, añade: «Se trata de familias que suelen ser descendientes de alguna persona que tuvo la enfermedad hace mucho tiempo y están muy distribuidas por toda España. A veces, incluso descendientes en América u otros lugares, dependiendo de las historias familiares. Muchas veces no existe un parentesco directo, pero generaciones atrás sí lo tuvieron».
La dificultad del diagnóstico
El diagnóstico de este tipo de patologías es difícil. «El problema viene más por el hecho de que el médico llegue a sospecharlo. Si no existen antecedentes familiares, bien porque no se conocen o porque se pensó que se trataba de otra enfermedad», apunta Izaro Kortazar Zubizarreta, neuróloga e investigadora del Hospital Universitario de Álava e IIS Bioaraba. «Los pacientes y sus familiares a veces se quejan de que han ido a muchos médicos. Como son enfermedades raras y comparten manifestaciones clínicas con otras enfermedades neurológicas y neurodegenerativas, normalmente cuando entran en contacto con un posible paciente no piensan en esta posibilidad. No es negligencia del sistema, sino que es difícil», concuerda Rodríguez.
La prueba analítica que confirma el diagnóstico es una punción lumbar. «Es invasiva y los especialistas solo recurren a ella cuando ya tienen una sospecha muy fuerte de que pueda ser ese tipo de enfermedad», indica Rodríguez. Si bien, Kortazar amplía: «A día de hoy se ha desarrollado la muestra del líquido cefalorraquídeo, una técnica que permite medir esa proteína anómala, detectar si está o no; eso hace años era impensable».
Sara González Navarro, vicepresidenta de la Fundación Española de Enfermedades Priónicas, subraya que el problema es que cuando aparecen esos síntomas y se solicitan esas pruebas, ya existen síntomas. «Significa que ya hay partes muy afectadas, el desarrollo es muy rápido. Se llaman enfermedades espongiformes porque dejan el cerebro con la forma de una esponja, los priones van matando las neuronas».
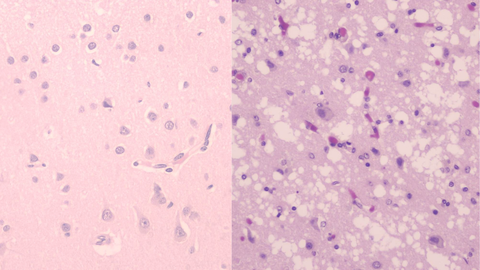
Los síntomas y la evolución de la enfermedad
Cada enfermedad priónica puede presentarse de manera diferente y, además, no todos los pacientes de esa patología cursan con los mismos signos clínicos. Los primeros síntomas de la enfermedad de Creutzfeldt-Jakob pueden ser del estado de ánimo, como cambios de humor, depresión, pérdidas de memoria, aislamiento social y falta de interés. En cuestión de semanas, se puede dar falta de coordinación e inestabilidad, mostrándose torpe. Puede progresar con alucinaciones, visión borrosa, rigidez y dificultades en el habla. Al poco tiempo, el paciente ya pierde sus facultades y requiere cuidados.
Si se trata de insomnio familiar fatal, los síntomas iniciales están relacionados con esa falta de sueño, que puede resultar en ataques de pánico, paranoias y fobias, que se vuelven más frecuentes con el transcurso de los meses, junto con una pérdida de peso acusada. En una etapa posterior aparece la demencia y, entre los 7 y los 36 meses desde el inicio de los primeros síntomas de la enfermedad, suele ocurrir el fallecimiento.
A diferencia de otras enfermedades degenerativas, «probablemente sea de las más rápidas en evolución, principalmente meses, si bien alguno puede tener una supervivencia un poco más larga», confiesa la neuróloga. Kortazar define dos caras de la enfermedad: «Hay quien inicia con problemas cognitivos, de memoria y lenguaje; mientras otros no pueden coordinar bien las extremidades, problemas de movimiento. Luego se solapan ambas y se dan todos estos signos».
Retos
El mayor reto a nivel clínico es conseguir una cura. «Principalmente para los casos familiares, porque los casos que son esporádicos es difícil hacer una intervención sobre algo que se da en una entre un millón de personas y que, generalmente, se detecta ya muy avanzado. Pero en los casos familiares jugamos con ventaja», comenta la neuróloga. Es posible saber qué personas pueden estar en riesgo a través de análisis genéticos en las familias donde se da un diagnóstico de este tipo. «En España existe una cierta distribución. Por ejemplo, hay más casos en País Vasco, Madrid y Galicia. Si hablamos de cuántas personas están en riesgo de pertenecer a estas familias, ahora mismo estamos llevando a cabo un estudio donde tenemos en torno a 200 personas. Luego habrá que ver ahí cuántos de los que tienen la mutación, lo desarrollan», añade.
Rodríguez recuerda sus primeros años investigando estas enfermedades priónicas: «Cuando empecé, en los dos mil, se sabía muy poco sobre ellas. Hemos avanzado mucho en la comprensión de los mecanismos de la enfermedad y nos estamos acercando a entenderlos de forma completa, pero no se da el mismo éxito a la hora de desarrollar tratamientos. Ese sería para mí el principal reto en este momento».
Unos avances que también han sido posibles gracias a la existencia de la Fundación Española de Enfermedades Priónicas. Se creó en mayo de 2013, gracias a la unión de una docena de personas. «A día de hoy, cada vez nos llegan más casos y antes. Las incertidumbres suelen ser cuánto tiempo puede quedar y qué es lo que va a pasar. Intentamos ponernos en la antesala de a qué se enfrentan y les ofrecemos muchos servicios que les puedan ayudar, tanto a nivel asistencial como legal. Somos conscientes de lo difícil que es enfrentarse a esto en soledad», concluye su vicepresidenta.
Comentarios
Publicar un comentario